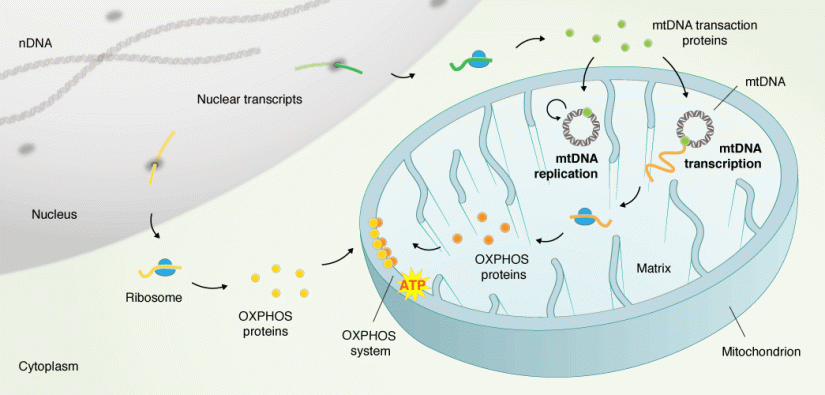
Mitochondria are needed for converting food into usable energy forms via the respiratory chain. Most cells contain hundreds to thousands of these organelles. Mitochondria have their own genomes (mtDNA) that in mammals encode 13 essential proteins of the respiratory chain. These genomes are continuously copied in order to maintain mtDNA copy number and meet cellular energy demands.
The existence of a separate mitochondrial genome is explained by the endosymbiotic theory, according to which the mitochondrion developed from ancient bacteria. During the course of time, most of the ancestral bacterial genes transferred into the host nuclear genome. All proteins necessary for mtDNA replication, as well as transcription and translation of mtDNA genes, are encoded in the nucleus. This explains why mitochondrial diseases can be caused by mutations in the mtDNA itself or in nuclear genes. Mutations in nuclear-encoded proteins required for mtDNA maintenance are an important cause of neurodegeneration and muscle diseases. Mitochondrial dysfunction is also associated with common age-associated diseases (e.g. diabetes mellitus type II and Parkinson disease).
In the laboratory we are investigating the basic mechanisms of human mtDNA replication and how this process is regulated. In addition, we address the functional consequences of disease-causing mutations in genes required for mtDNA maintenance. Much of the work is performed using in vitro biochemistry and reconstituted systems for mammalian mtDNA replication, using different types of artificial templates and recombinant proteins. We complement this approach with cell-based assays and structural biology. We collaborate extensively with clinicians working with mitochondrial disorders and experts in mouse genetics.
